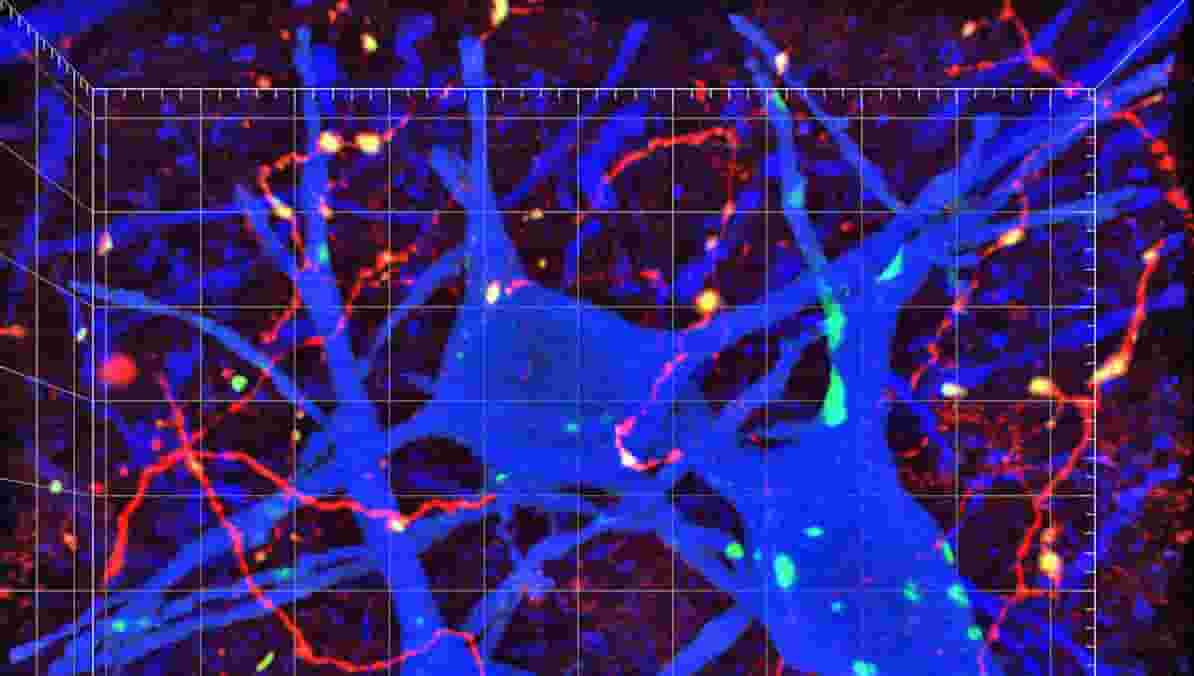
Strengthening and weakening the connections between neurons, known as synapses, is vital to the brain’s development and everyday function. One way that neurons weaken their synapses is by swallowing up receptors on their surfaces that normally respond to glutamate, one of the brain’s excitatory chemicals.
In a new study, MIT neuroscientists have detailed how this receptor reabsorption takes place, allowing neurons to get rid of unwanted connections and to dampen their sensitivity in cases of overexcitation.
“Pulling in and putting out receptors is a dynamic process, and it’s highly regulated by a neuron’s environment,”
says Elly Nedivi, a professor of brain and cognitive sciences and member of MIT’s Picower Institute for Learning and Memory.
“Our understanding of how receptors are pulled in and how regulatory pathways impact that has been quite poor.”
Nedivi and colleagues found that a protein known as CPG2 is key to this regulation, which is notable because mutations in the human version of CPG2 have been previously linked to bipolar disorder. “This sets the stage for testing various human mutations and their impact at the cellular level,” says Nedivi, who is the senior author of a Current Biology paper describing the findings.
The paper’s lead author is former Picower Institute postdoc Sven Loebrich. Other authors are technical assistant Marc Benoit, recent MIT graduate Jaclyn Konopka, former postdoc Joanne Gibson, and Jeffrey Cottrell, the director of translational research at the Stanley Center for Psychiatric Research at the Broad Institute.
Forming a Bridge
Neurons communicate at synapses via neurotransmitters such as glutamate, which flow from the presynaptic to the postsynaptic neuron. This communication allows the brain to coordinate activity and store information such as new memories.
Previous studies have shown that postsynaptic cells can actively pull in some of their receptors in a phenomenon known as long-term depression (LTD).
This important process allows cells to weaken and eventually eliminate poor connections, as well as to recalibrate their set point for further excitation. It can also protect them from overexcitation by making them less sensitive to an ongoing stimulus.
Pulling in receptors requires the cytoskeleton, which provides the physical power, and a specialized complex of proteins known as the endocytic machinery.
This machinery performs endocytosis — the process of pulling in a section of the cell membrane in the form of a vesicle, along with anything attached to its surface. At the synapse, this process is used to internalize receptors.
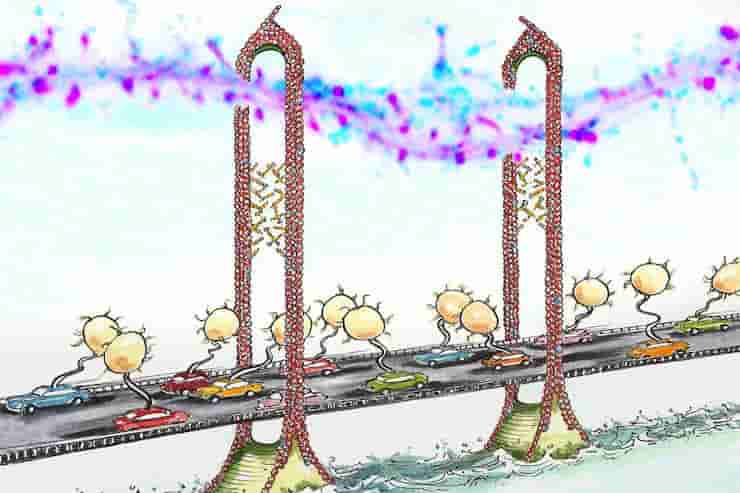
Until now, it was unknown how the cytoskeleton and the endocytic machinery were linked. In the new study, Nedivi’s team found that the CPG2 protein forms a bridge between the cytoskeleton and the endocytic machinery.
“CPG2 acts like a tether for the endocytic machinery, which the cytoskeleton can use to pull in the vesicles,” Nedivi says. “The glutamate receptors that are in the membrane will get pinched off and internalized.”
They also found that CPG2 binds to the endocytic machinery through a protein called EndoB2. This CPG2-EndoB2 interaction occurs only during receptor internalization provoked by synaptic stimulation and is distinct from the constant recycling of glutamate receptors that also occurs in cells.
Nedivi’s lab has previously shown that this process, which does not change the cells’ overall sensitivity to glutamate, is also governed by CPG2.
“This study is intriguing because it shows that by engaging different complexes, CPG2 can regulate different types of endocytosis,”
says Linda Van Aelst, a professor at Cold Spring Harbor Laboratory who was not involved in the research.
When synapses are too active, it appears that an enzyme called protein kinase A (PKA) binds to CPG2 and causes it to launch activity-dependent receptor absorption. CPG2 may also be controlled by other factors that regulate PKA, including hormone levels, Nedivi says.
Link to Bipolar Disorder
In 2011, a large consortium, including researchers from the Broad Institute, discovered that a gene called SYNE1 is number two on the hit list of genes linked to susceptibility for bipolar disorder. They were excited to find that this gene encoded CPG2, a regulator of glutamate receptors, given prior evidence implicating these receptors in bipolar disorder.
In a study published in December, Nedivi and colleagues, including Loebrich and co-lead author Mette Rathje, identified and isolated the human messenger RNA that encodes CPG2. They showed that when rat CPG2 was knocked out, its function could be restored by the human version of the protein, suggesting both versions have the same cellular function.
Rathje, a Picower Institute postdoc in Nedivi’s lab, is now studying mutations in human CPG2 that have been linked to bipolar disorder. She is testing their effect on synaptic function in rats, in hopes of revealing how those mutations might disrupt synapses and influence the development of the disorder.
Nedivi suspects that CPG2 is one player in a constellation of genes that influence susceptibility to bipolar disorder.
“My prediction would be that in the general population there’s a range of CPG2 function, in terms of efficacy,” Nedivi says. “Within that range, it will depend what the rest of the genetic and environmental constellation is, to determine whether it gets to the point of causing a disease state.”
Reference:
- Sven Loebrich, Marc Robert Benoit, Jaclyn Aleksandra Konopka, Jeffrey Richard Cottrell, Joanne Gibson, Elly Nedivi. CPG2 Recruits Endophilin B2 to the Cytoskeleton for Activity-Dependent Endocytosis of Synaptic Glutamate Receptors. Current Biology DOI: 10.1016/j.cub.2015.11.071
Original Article by Anne Trafton. Republished courtesy of MIT News
Last Updated on November 28, 2023